Publication Summary
A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization
1 Department of Cell Biology, Harvard Medical School, Boston, MA; 2 Laboratory of Systems Pharmacology, Harvard Medical School, Boston, MA; 3 Cell Signaling Technologies, Danvers, MA; 4 These authors contributed equally.
Mol Cell (2017) 65(2):361-370.
doi:10.1016/j.molcel.2016.12.005 / PMID:28065596 / PMCID:PMC5250569
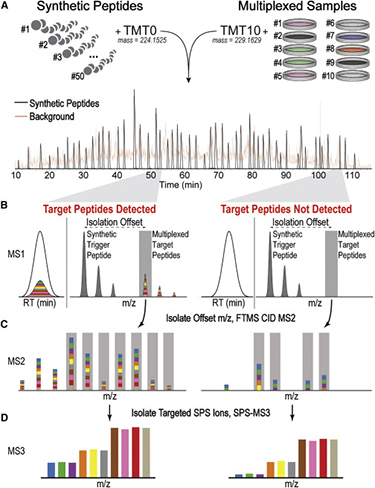
The 10-plex TOMAHAQ Workflow
(A) Spiked-in internal standards are mixed with up to ten multiplexed endogenous proteomes and combined before analysis.
(B) This results in two precursor ion clusters that have identical elution profiles. Synthetic trigger and multiplexed target peptides are separated by a known mass offset. The presence of synthetic trigger peptides is used to prompt selection (gray bar) and quantitative analysis of target peptides at all precursor abundances, obviating the need for target detection in the MS1.
(C) MS2 analysis of target peptides is used to select b- and y-type fragment ions for synchronous precursor selection (SPS, gray bars).
(D) Targeting interference-free fragment ions yields accurate quantitation in an MS3 scan, even if multiple interfering peptides are co-isolated.
Synopsis
In proteomics, two fundamental types of multiplexing are available to increase throughput. Many peptides can be targeted for quantification during a single run (peptide multiplexing), and higher-order multiplexing is possible at the sample level using a method such as isobaric labeling. Targeted mass spectrometry-based analyses have become vital for multiplexing the measurement of many peptides, but isobaric tagging-based multiplexing in targeted assays has not been demonstrated with unfractionated mixtures. Thanks to the evolution of mass spectrometry instrumentation capable of sensitive MS3 analysis of reporter ions, we here demonstrate that 10-plex sample multiplexing with accurate, targeted quantification is possible directly from proteolyzed cell lysates.
Key Findings
- Combined peptide and sample multiplexing increases throughput for targeted analyses.
- Unfractionated targeted 2D multiplexing reproduces global proteome quantitation.
- This method allowed monitoring of 69 proteins across 180 cancer cell lines in 48 hr (16 min/sample).
- NCI-60 profiling revealed a correlation between BAZ1B expression and doxorubicin response.
Abstract
Targeted mass spectrometry assays for protein quantitation monitor peptide surrogates, which are easily multiplexed to target many peptides in a single assay. However, these assays have generally not taken advantage of sample multiplexing, which allows up to ten analyses to occur in parallel. We present a two-dimensional multiplexing workflow that utilizes synthetic peptides for each protein to prompt the simultaneous quantification of >100 peptides from up to ten mixed sample conditions. We demonstrate that targeted analysis of unfractionated lysates (2 hr) accurately reproduces the quantification of fractionated lysates (72 hr analysis) while obviating the need for peptide detection prior to quantification. We targeted 131 peptides corresponding to 69 proteins across all 60 National Cancer Institute cell lines in biological triplicate, analyzing 180 samples in only 48 hr (the equivalent of 16 min/sample). These data further elucidated a correlation between the expression of key proteins and their cellular response to drug treatment.
Explore the data
We encourage readers to explore the method and the data generated as part of this study through the associated resources listed below.
Available resources and data
| Method | Summary webpage for the Triggered by Offset Mass Accurate-mass High-resolution Accurate Quantitation (TOMAHAQ) method | Summary |
| Tutorial | Tutorial for setting up the TOMAHAQ method in the method editor of your mass spectrometry instrument | Tutorial |
| Data | NCI-60 data browser | Browser |
Funding Sources
The Charles A. King Trust (M.W.) and NIH grants DK098285 (J.A.P.), GM67945 (S.P.G.), P50 GM107618 (R.A.E. and A.R.E.), U54 HL127365 (R.A.E. and A.R.E.), and R01GM103785 (M.W.).